摘要:在生物的基因序列中,蕴含了其所有的特点和规律,是大自然隐藏在生物千变万化形态下的“密码”。在本文中我们选取21种HIV病毒基因数据,应用基于非序列比对的平均互信息方法提取出它们的序列特征,结合相关系数和离差平方和方法(Ward法)对其进行分类。此种分类方法有别于传统的序列对比方式,运算简单,速度快捷且得到了合理的分类结果。
关键词:平均互信息;基因组;离差平方和方法
引言
在医学领域,从DNA分子水平来研究疾病的起因发展与分类,解读病毒基因的“密码”,正日益引起分子生物学者、数学、计算机以及信息网络科学研究人员的重视。如何分析这些DNA序列数据,提取出能够量化的“信息”来描述它们之间的联系,是当前研究的热门问题。HIV病毒在进化过程中形成了三种亚型分类,目前对这种进化分类常用的方法有最大简约法、距离矩阵法和最大似然法等。相应的也有一系列软件,如:PHYLIP、PAUP和MEGA等。通常在应用这些方法之前,都要对序列进行比对(sequence alignment),常用的软件有CLUSTRALW等。
本文提取不同DNA序列的平均互信息(Average Mutual Information,AMI)[2]作为特征参数,构造AMI向量,通过AMI向量的相关系数定义不同DNA序列之间的距离,利用离差平方和法对距离矩阵进行聚类分析,从而得到他们的进化关系。此种方法是非序列比对方法,计算简单且速度较快,对大量数据的处理非常方便,在医学领域中有着广泛的应用。
1.理论与方法
1.1平均互信息(AMI)
DNA序列是4种核苷酸A、C、G、T的集合,如果x代表在基因序列上某一位置的核苷酸,则y为在x下游方向间隔k个位置的核苷酸。n\-k(x,y)表示核苷酸x其下游间隔k个位置为y的组合的个数,这样就P\-k(x,y)表示核苷酸x其下游间隔k个位置为y的条件概率。p(x)和p(y)分别是基因序列中核苷酸x和y的概率。
当选取k=0时,就表示了紧邻二联体核苷酸的关联程度,k=1时表示次紧邻二联体核苷酸的关联程度。[3]i\-k就是基因序列的平均互信息(AMI),不同的k值对应不同的i\-k,对于每一基因组,我们都能够得到一组数据i\-0,i\-1,…,i\-k,从而构成向量I=
(i\-0,i\-1,…,i\-k),不同的基因序列,可以得到不同的向量I,J,L。
1.2 相关系数
在本文中我们使用的是线性相关系数,它反了映两个数据集之间的线性相关程度。若相关系数为,表示两个数据集之间呈现完美的正线性相关;若相关系数为,则表示量数据集之间是负线性相关;若相关系数为0,则表示两组数据集之间没有线性相关性。
1.3 聚类分析
我们通过计算不同物种两两之间的AMI距离,可以得到不同物种之间的距离从而得到一个距离矩阵。对于这个矩阵,本文选用离差平方和方法进行聚类。
2.基因数据与结果讨论
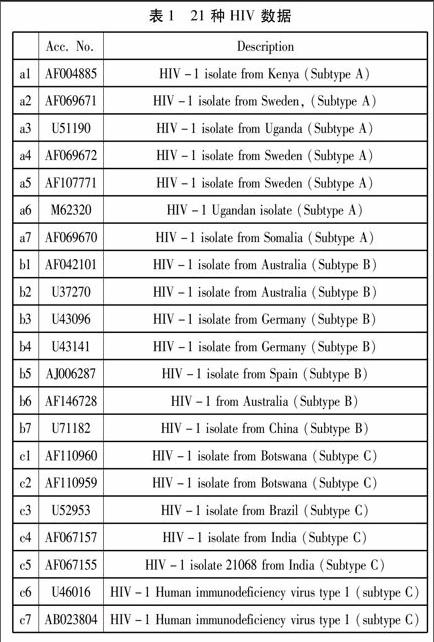
2.1 基因数据:21种HIV 数据
21种HIV病毒基因分为三种亚型,用a、b、c分别表示,每种又各有七种,数据来自NCBI(http://www.ncbi.nlm.nih.gov)。这21种HIV数据,长度比较一致,都在10000个核苷酸上下。
2.2 数据计算
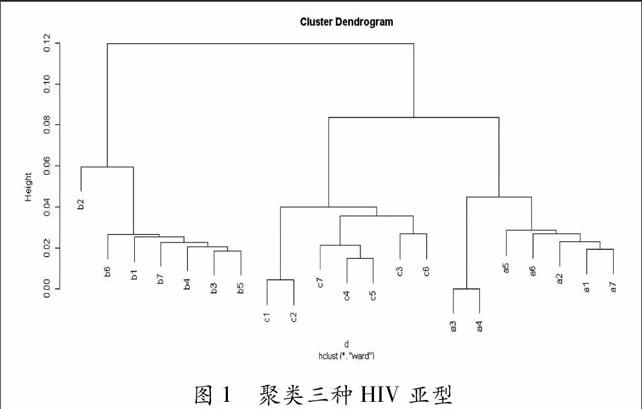
对于这21种HIV病毒,首先提取各自序列的AMI向量,每种病毒得到一个向量 。对于参数k的选取,我们选取了10、50、100、200、300、400、500、600、700等多个值,综合各向量之间的距离和聚类分析的结果来看,k取500是比较合适的,k过小会丢失基因组的一些关联信息,k过大对结果没有什么影响,这样AMI向量共有501个分量。对于这21个向量计算两两之间的相关系数,从而组成一个距离矩阵。我们将这个距离矩阵输入R软件,使用离差平方和法进行聚类分析,得到分类结果如图1所示:
3.结果分析
在Mark等人[4]的文章中,也对这组数据做了分析,Mark等使用UPGMA tree、2维和3维图等方法对这组数据做了分析,将这21种HIV病毒分成了三类。在本文中我们使用R软件,应用离差平方和法更为简单方便,计算速度更快,由图1可以看出同样对这21种HIV病毒做了很好的区分,将其分为a、b、c三类,达到完全区分的目的。
[参考文献]
孙啸,陆祖宏,谢建明.生物信息学基础[M].北京:清华大学出版社,2005:238-239.
[2] Mark Bauer,Sheldon M Schuster and Khalid Sayood.The Average Mutual Information Profile as a Genomic Signature[J].BMC Bioinformatics 2008,9:48 doi:10.1186/1471-2105-9-48.
[3] 罗辽复.生命进化的物理观[M].上海:上海科学技术出版社,2000,168-183.
[4] Ouyang Z,Zhu H,Wang J,et al.Multivariate entropy distance method for p rokaryotic gene identification [J]. J Bioinform ComputBiol, 2004,2(2):353-73.
(作者单位:武警山东省总队训练基地,山东 济南 250000)endprint